Project Z01
Z01 MHC ligands and T cell epitopes
Dr. Andreas Schlosser, Würzburg
We use mass spectrometry (MS) for analyzing the MHC class I and class II peptide repertoire (immunopeptidome) (Fig. 1). In order to extend the mass spectrometry-detectable landscape of the immunopeptidome we recently introduced the application of restricted access material (RAM) for MHC peptide sample preparation. This enables the identification of highly hydrophobic peptides, which previously escaped MS-detection. Actually, we are developing novel methods for improving the sensitivity and identification rate of MHC peptides by chemical modification.
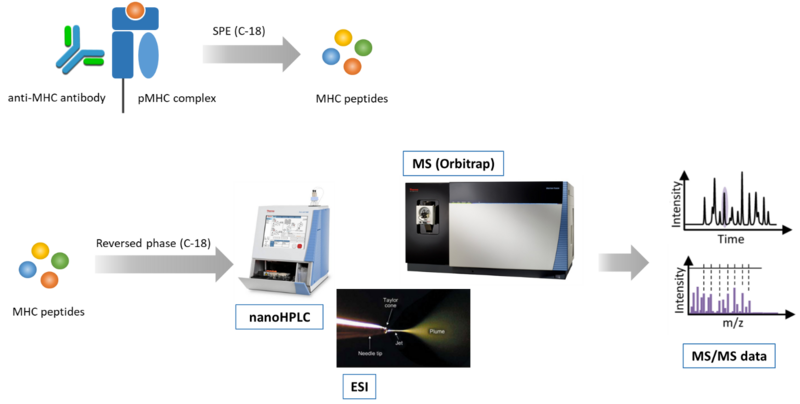
Fig. 1: Mass spectrometry-based immunopeptidomics workflow. A) Peptide-loaded MHC complexes (pMHC) are isolated by affinity purification with specific antibodies (e.g. anti-panHLA class I antibody W6/32), and MHC peptides are isolated by solid phase extraction (SPE). B) MHC peptides are separated by reversed-phase chromatography, ionized by electrospray, and analyzed by tandem mass spectrometry (MS/MS).
In close collaboration with Florian Erhard (P01) we have developed a novel computational approach termed Peptide-PRISM for the identification of MHC-I peptides from mass spectrometric data (Fig. 2) [1]. Compared to conventional database search engines that are typically used for MHC peptide identification, Peptide-PRISM shows significantly higher peptide identification rates. In addition, Peptide-PRISM enables the fast and reliable identification of cryptic MHC peptides. These peptides are derived from allegedly non-coding regions of the genome, such as the 5’-UTR or 3’-UTRs of mRNAs, or from ncRNAs. We found that about 25% of HCMV-derived MHC-I peptides derive from open reading frames (ORFs) annotated as non-coding, and are thus classified as cryptic, viral MHC peptides. The immunological role of these MHC peptides is actually unknown, and currently under investigation.

Fig. 2: Peptide-PRISM, a novel tool for efficient and reliable identification of MHC peptides. Peptide-PRISM combines de novo peptide sequencing with fast database matching, enabling searches in ultra-large databases, such as the 6-frame translated human genome. A stratified FDR filtering strategy using a target-decoy approach enables reliable control of FDR (Figure from [1]).
We have established metabolic stable isotope labeling (pulsed SILAC) as well as isobaric chemical labeling (TMT, iTRAQ). This enables us to study dynamic changes within immunopeptidomes, e.g. upon viral infection or expression of immunoevasins. Due to its multiplexing capabilities, TMT labeling can be used for time course experiments. In addition, we apply dynamic SILAC to measure the turnover of MHC peptides.
In collaboration with P03, we are analyzing the immunopeptidome of HLA-E with and without viral infection. In collaboration with P04, we are optimizing the analyses immunopeptidomes of soluble HLA molecules (sHLA).
References
[1] Erhard, F.; Dölken, L.; Schilling, B.; Schlosser, A. Identification of the cryptic HLA-I immunopeptidome. Cancer Immunol. Res. 2020, 8, 1018–1026, doi:10.1158/2326-6066.CIR-19-0886.