Project P02
P02 Solving the m04 paradox: Evasion of missing-self recognition and CD8 T cell killing by MAT uORF
Dr. Vanda Juranic Lisnic & Prof. Dr. Stipan Jonjic PhD
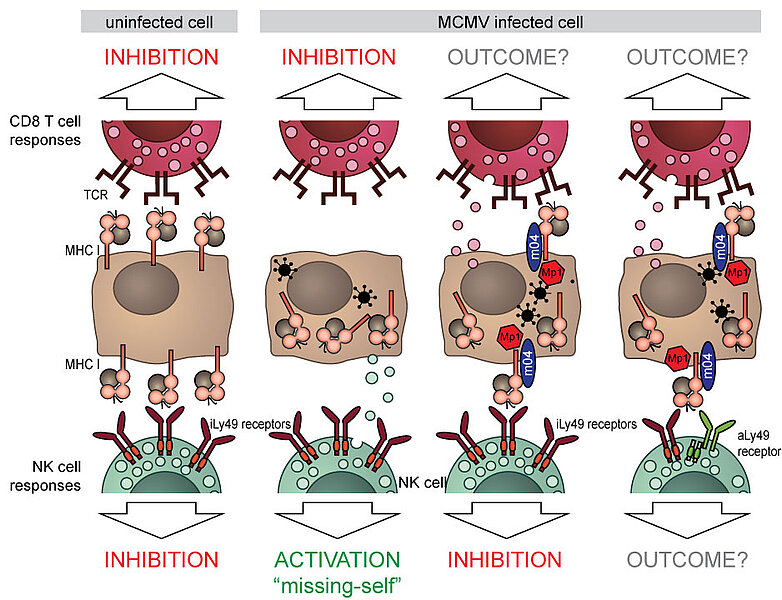
Cytomegaloviruses (CMVs) are expert evaders of nearly every aspect of our immune system. One of these strategies is the downregulation of surface MHC I molecules (major histocompatibility molecules class I). As MHC I molecules present peptides (small fragments) of all proteins generated within the cells to cytotoxic T cells, downregulation of MHC I molecules is an effective way of hiding a viral presence inside the cell from T cells.
Natural killer (NK) cells, lymphocytes that belong to innate immune systems, possess receptors called Ly49 in mice or KIR in humans. These receptors recognize MHC I molecules on the cell surface and inhibit the NK cell, preventing it from uncontrolled killing of cells. The NK cell is thus said to recognize “self” molecules. When MHC I molecules are downregulated upon virus infection, the NK cell is no longer inhibited by KIR or Ly49 receptors. In particular, when there are other activating signals or inflammation, the NK cell will kill the infected cell with downregulated MHC I molecules in a process termed “missing-self” killing. To prevent this, mouse cytomegalovirus (MCMV) have evolved to express the m04 protein, which escorts certain types of MHC I molecules to the cell surface in order to engage inhibitory Ly49 receptors and prevent “missing-self” killing. Interestingly, these m04 only interact with those MHC I molecules that can engage inhibitory Ly49 receptors. Even more interesting is the fact that although the number of m04-escorted MHC I molecules on the cell surface is significantly lower compared to the levels on uninfected cells, modified MHC I molecules still inhibit missing-self recognition and killing with similar efficacy.
During the millennia of our co-evolution with herpesviruses, murine immune systems have adapted to this strategy and developed activating NK cell receptors that can recognize these altered-self MHC I molecules. We recently identified that 3 activating Ly49 receptors recognize m04-rescued MHC I molecules. Furthermore, we identified a novel small viral protein (MATp1), which binds to MHC-I in concert with m04 to rescue MHC-I surface expression.
In the course of this project, we will investigate molecular mechanisms in which m04 and MATp1 collaborate to bring back certain types of MHC I molecules and explore the effect that this has on the virus control mediated by various components of the immune system.